
問醫D
罕病不可怕 : 深入認識法布瑞氏症,及早篩查增療效!

李沛威醫生
心臟科專科醫生
2026/07/03
經常感到手腳灼熱疼痛、運動後特別疲倦,甚至在炎熱天氣下也難以正常排汗?這些症狀看似源於壓力或體質問題,其實可能與一種罕見遺傳代謝疾病——法布瑞氏症(Fabry disease)有關。1 這是一種 X 染色體連鎖遺傳疾病,因體內酵素缺乏導致醣脂逐漸積聚於心臟、腎臟及神經系統,長遠可能損害多個器官。1 由於疾病表現多樣且缺乏特異性,早期症狀與常見疾病相似,容易延誤診斷。1 因此,及早識別高危人士並進行適當檢測至關重要。1 為了幫助大家更了解此病,心臟科專科醫生李沛威醫生將深入講解法布瑞氏症的成因、臨床表現及不同的治療方案。
法布瑞氏症(Fabry disease)是什麼疾病?
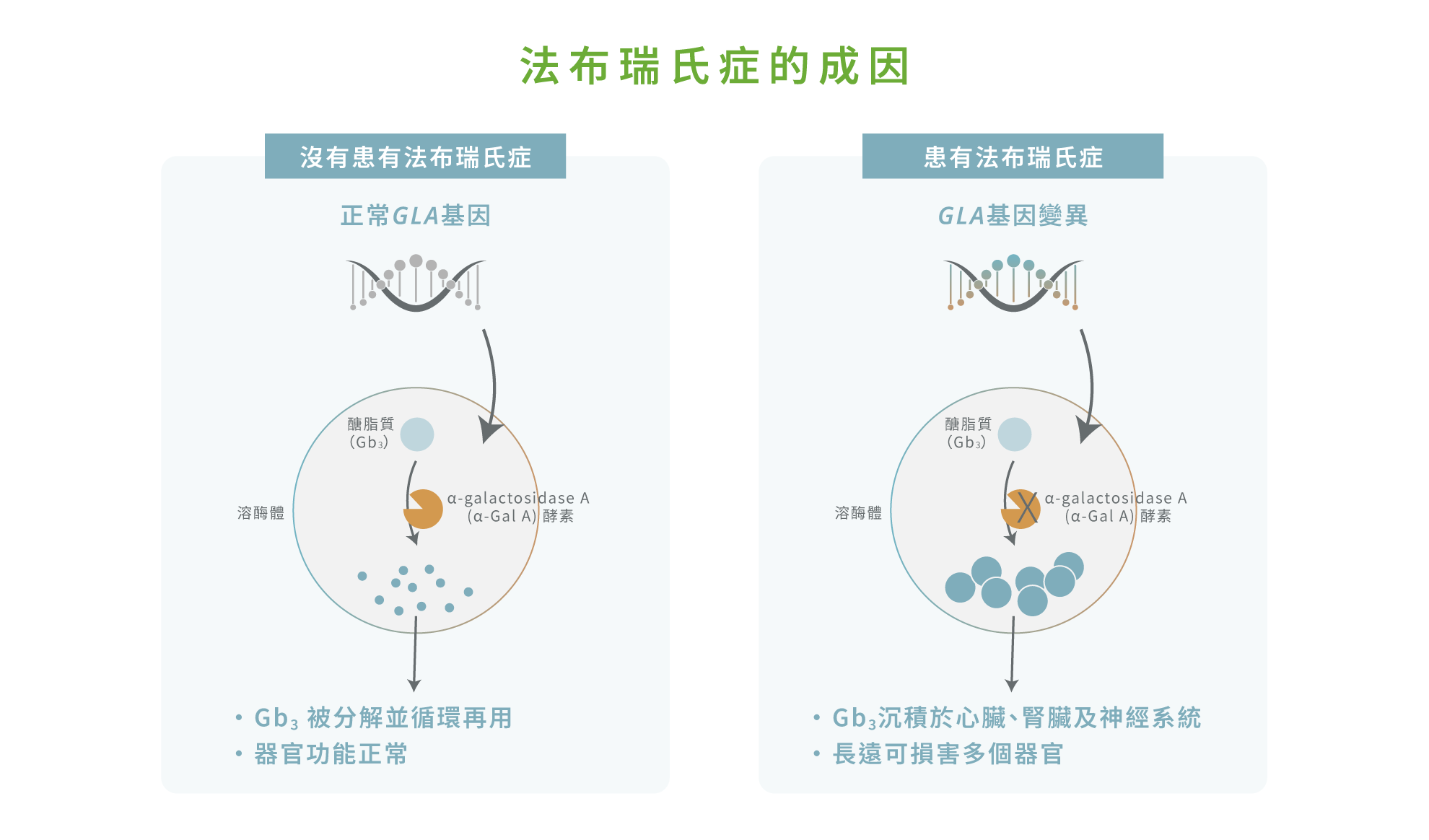
法布瑞氏症(Fabry disease)是一種先天性遺傳代謝疾病,屬於溶小體儲積症(Lysosomal storage disorder)。1,2 這種疾病以 X 染色體連鎖遺傳(X-linked inheritance)為特徵,由 GLA 基因突變導致體內α-galactosidase A (α-Gal A) 酵素活性不足或缺失,從而造成醣脂質(Globotriaosylceramide; Gb3)代謝異常。2 未被分解的 Gb3 及其衍生物會逐漸沉積於血管內皮細胞、腎臟、心肌細胞、神經系統及皮膚組織,造成慢性進行性器官功能障礙。2,3
由於法布瑞氏症是遺傳病,患者是否很大機會將病症傳給下一代?患者的家族成員應否接受檢測?
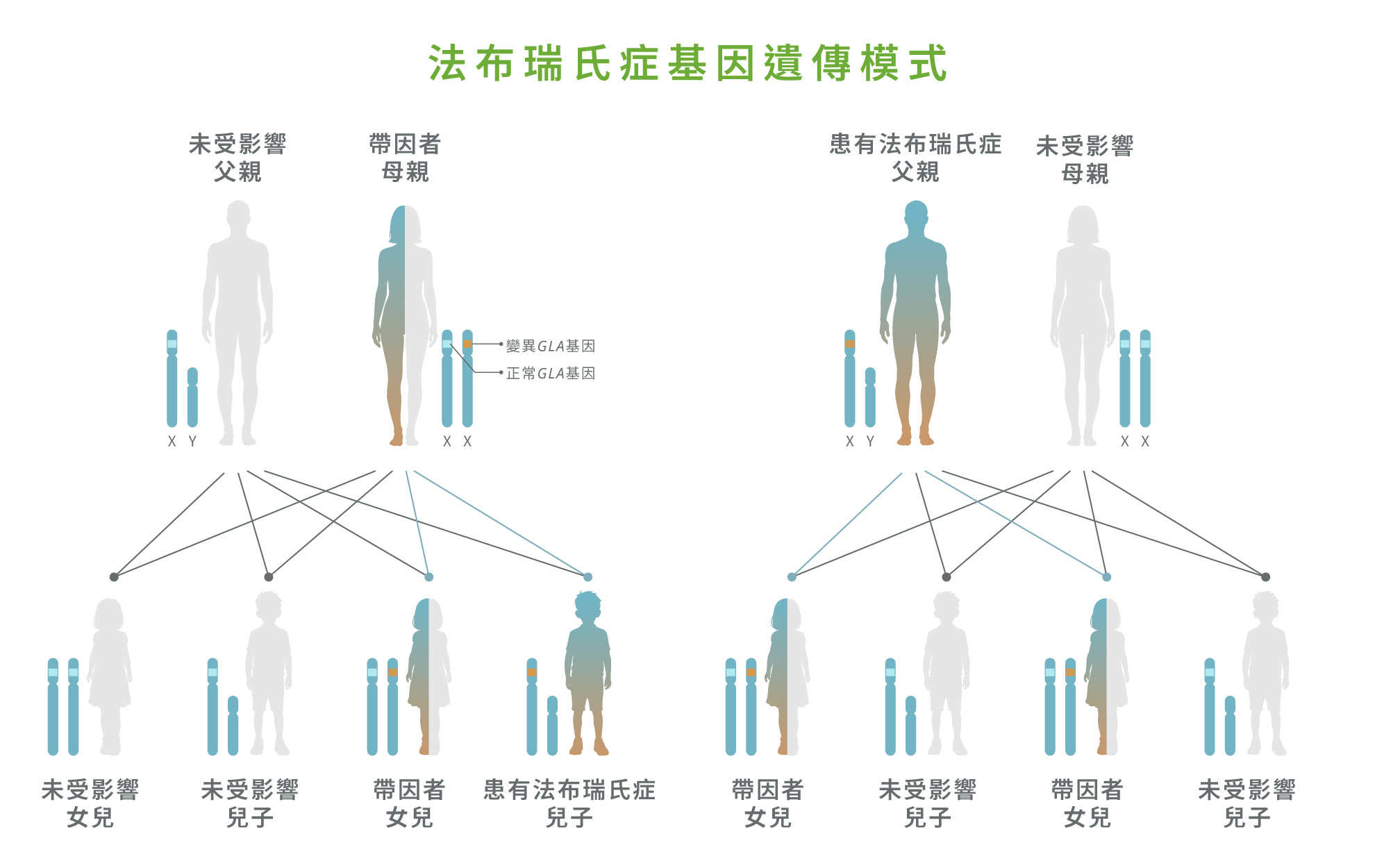
法布瑞氏症屬 X 染色體連鎖遺傳疾病。1 根據遺傳模式,若母親為帶因者,每名子女都有 50% 機會遺傳到致病基因;若父親為患者,其女兒便會成為帶因者,而兒子則不會受影響。1 鑑於法布瑞氏症可能長期缺乏明顯症狀,國際指引一致建議對確診患者的近親進行家族連鎖篩查(Cascade family screening)。1,2 研究顯示,每確診一名指標病例,平均可額外識別至少五名受影響家族成員,當中不少患者於疾病早期階段得到診斷,從而在器官損害前及早介入治療,顯著改善治療效果。1,2 此外,家族篩查可幫助患者及其家屬了解遺傳風險,作出生育決策,並在需要時考慮基因諮詢、產前診斷或其他生殖選擇,以降低將疾病傳遞給下一代的可能性。2
哪些早期症狀較容易被忽視且與其他疾病混淆?
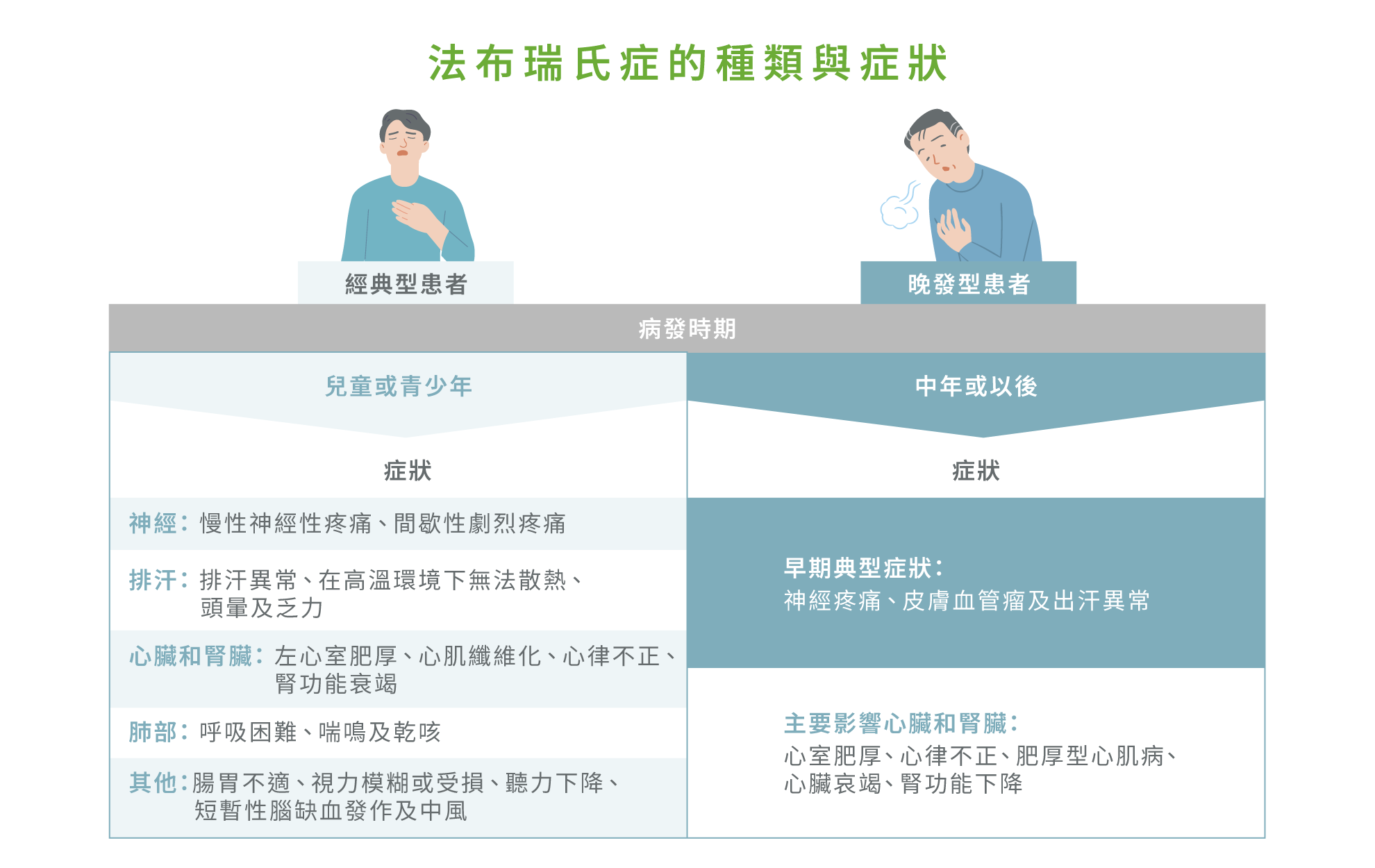
法布瑞氏症可分為經典型(Classical type)和成人晚發型(Later-onset type),兩者在性別上有明顯差異。2 經典型患者 α-Gal A 酵素活性幾乎完全缺失(<1% 正常平均值),導致GL-3 顯著堆積於血管內皮細胞、心肌細胞、平滑肌細胞及腎小球足細胞,症狀多於兒童或青少年期出現,且疾病可能影響多個器官,導致嚴重併發症。2,4 典型早期症狀包括慢性神經性疼痛、間歇性劇烈疼痛、腸胃不適及視力模糊或受損等。2 患者也可能會出現排汗異常,在高溫環境下無法散熱,伴隨頭暈及乏力等症狀,但這些症狀往往未被及時聯想到為遺傳疾病,導致診斷延誤。2 隨年齡增長,患者亦可能會出現左心室肥厚、心肌纖維化、心律不正、腎功能衰竭、聽力下降、短暫性腦缺血發作及中風;肺部問題包括呼吸困難、喘鳴及乾咳。2
而成人晚發型患者,由於α-Gal A酵素活性仍有部分保留,症狀出現年齡及臨床表現差異較大,從輕微至嚴重不等。2,4 成人晚發型法布瑞氏症主要影響心臟及腎臟,多於中年或更晚時期出現典型心臟症狀,包括左心室肥厚、心律不正及心臟磁力共振影像異常,部分患者亦可能會出現腎功能下降。2 女性患者的臨床表現差異更大,可呈無症狀、僅影響少數器官,或表現與經典型男性相似的嚴重症狀,這取決於致病突變類型及 X 染色體隨機失活(Lyonization)情況。2
在香港,成人晚發型法布瑞氏症最常見的致病原因是c.640‑801G>A致病變異(原稱 IVS4+919G>A 基因突變)。5 此類患者早期典型症狀(如神經疼痛、皮膚血管瘤及出汗異常)較少,主要以心臟症狀為主(如左心室肥厚、肥厚型心肌病等),導致早期臨床識別較為困難。5,6 患者通常在出現了心室肥厚、心臟衰竭或腎功能衰退後才被確診,且因左心室肥厚為主要症狀,患者容易被誤診為高血壓性心臟病或肥厚型心肌病。7-10
為什麼這個疾病難以確診?有哪些檢查能有效檢測此病?
法布瑞氏症由於屬罕見疾病,臨床認知不足,症狀多樣且缺乏特異性,常被誤診為其他常見疾病,導致診斷延誤。11 研究指出,從症狀出現到確診的平均診斷延誤時間至少為 3 年,部分患者甚至超過 20 年。11
成人晚發型患者多表現為左心室肥厚,容易被誤認為高血壓性心臟病或肥厚型心肌病,進一步延誤正確診斷。7,10 此外,法布瑞氏症在神經系統表現上亦可能與多發性硬化症相似,例如反覆出現的肢體麻木、刺痛、灼熱感、頭暈、疲倦、熱不耐受等,臨床上容易混淆。12 部分患者的腦部影像檢查亦可能會出現類似多發性硬化症的病灶,使診斷更加困難,容易長期被誤診或未能及早辨識。12
目前,法布瑞氏症的檢測方法主要包括 : 2,13-16
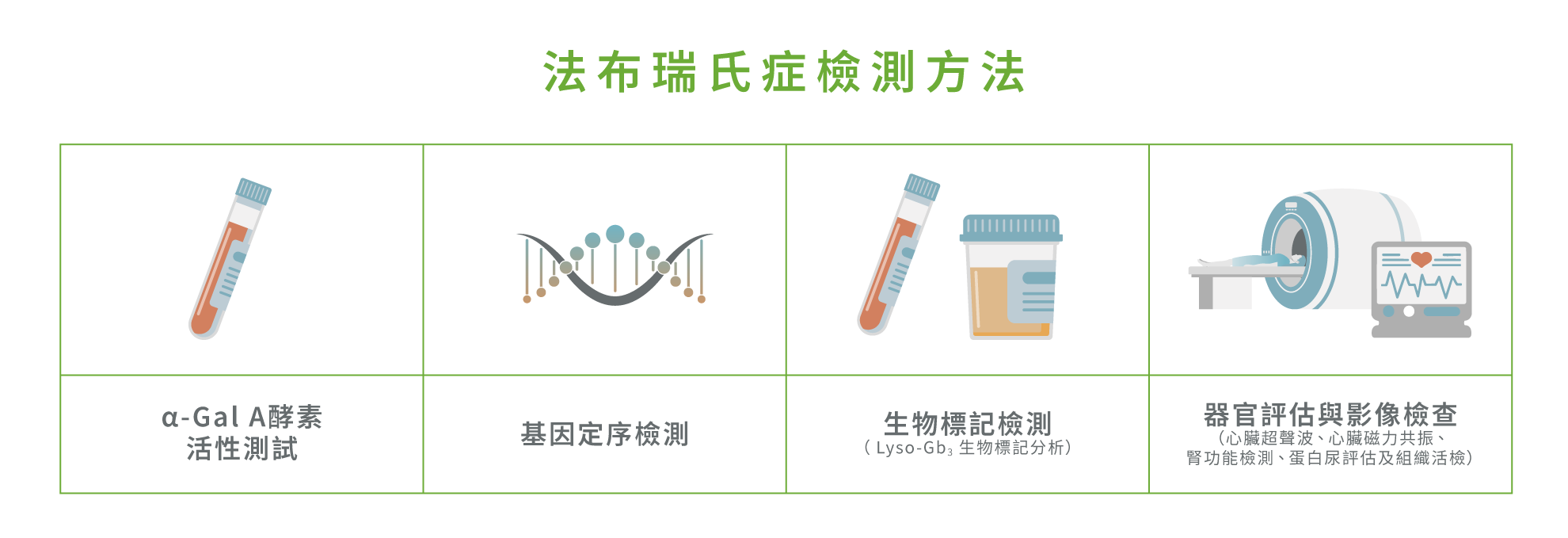
酵素活性測試: α-Gal A酵素活性測試主要用於男性患者的初步篩查與診斷。2 當其活性降至正常值1% 或以下,便可高度懷疑甚至確診法布瑞氏症。13,14 然而,該檢測對女性患者的診斷敏感度不足,原因在於女性體內存在 X 染色體隨機失活(Random X-chromosome inactivation) 現象,即使攜帶致病突變,其 α-Gal A 酵素活性仍可能呈現正常水平或僅輕微下降。因此,酵素測試容易漏診,不適合作為單一確診依據,必須進一步進行基因層面的確認1,2,14
基因定序檢測:GLA 基因定序檢測被視為適用於男女患者確診的黃金標準,能直接鑑定致病突變,對女性患者尤為關鍵,有助於避免漏診並提升整體診斷準確性1,14
生物標記檢測:血液或尿液中的 Lyso-Gb3(去醣基球三糖神經醯胺)生物標記測試則可反映體內醣脂的累積程度,作為輔助診斷工具及疾病活動度評估,有助於辨識 α-Gal A 活性正常或邊緣正常的女性患者。15 測試亦可用於疾病分層、治療決策評估及療效追蹤15
器官評估與影像檢查 : 心臟超聲波、心臟磁力共振、腎功能檢測、蛋白尿檢測能評估是否必須進行組織活檢,可顯示心臟肥厚、心肌纖維化、腎小球病變等典型病理變化,作為評估多個器官受影響程度及疾病進展的重要輔助工具,但並非單獨用於確診2
人工智能輔助影像分析:近年研究亦指出,人工智能輔助心臟超聲波影像分析可協助區分法布瑞氏症與其他心肌肥厚疾病,作為篩查工具有助提升早期診斷效率,但目前尚未廣泛應用於臨床常規診斷16
目前法布瑞氏症有哪些治療方案?現時有哪些藥物是已列入醫管局的藥物名冊供患者使用?
現時,國際認可的法布瑞氏症治療主要包括兩大核心策略 :
- 酵素替代治療(Enzyme replacement therapy; ERT)是法布瑞氏症的標準治療之一,透過補充患者體內缺乏的酵素進行治療,適用於男女確診患者。1
- 口服伴護小分子療法(Pharmacological chaperone therapy; PCT):適用於特定基因突變的患者。1
上述治療藥物均已納入香港醫院管理局藥物名冊,供合資格患者在專科醫生評估或基因檢測後使用。17,18
如果未及時接受適當治療,法布瑞氏症可能導致哪些嚴重後果?
若未接受適當治療,法布瑞氏症患者體內的醣脂將持續累積,最終導致不可逆轉的多種器官損害,主要影響心臟、腎臟、腦部及周邊神經系統 : 2
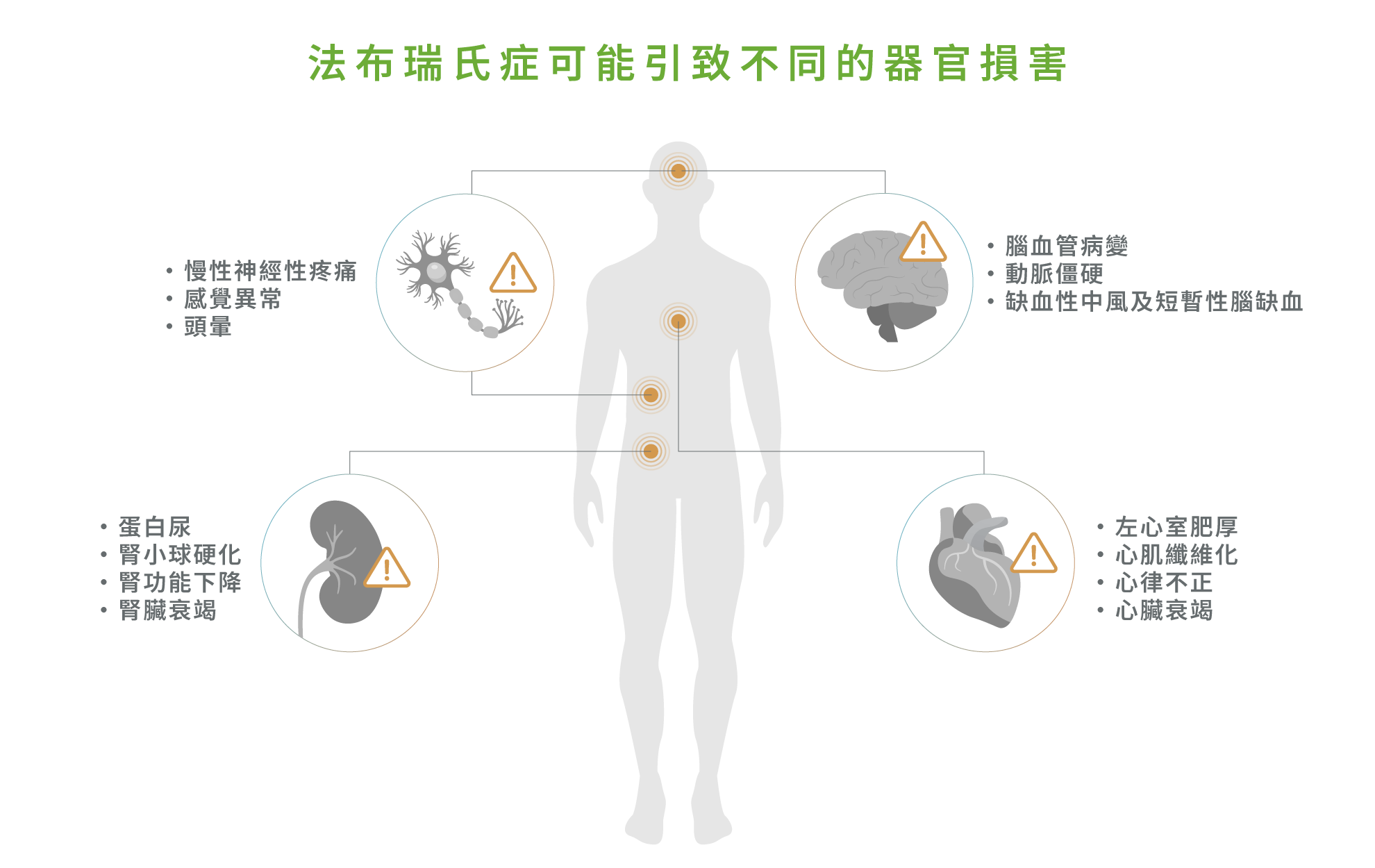
心臟:長期醣脂堆積會引致左心室肥厚,隨後可能形成心肌纖維化,進而導致心律不正(如房顫)及心臟衰竭,並增加猝死風險1,2
腎臟:患者早期可能會出現蛋白尿,隨著疾病進展,腎小球逐漸硬化並出現腎功能下降及慢性腎功能衰竭。嚴重患者最終可能需接受透析或腎移植1,2
腦血管系統:醣脂沉積可引起腦血管病變及動脈僵硬(如主動脈僵硬),進而增加缺血性中風及短暫性腦缺血發作的風險1,2
周邊神經系統:長期受影響則可導致慢性神經性疼痛、感覺異常(如對溫度敏感度下降)、頭暈,進而導致生活功能下降 1,2
雖然法布瑞氏症需要長期管理,患者應如何與疾病共存?及早治療對病情有何幫助?
儘管法布瑞氏症需要長期且終身管理,患者仍可以通過接受適當治療來與疾病共存。早期診斷與及時介入對於延緩病情惡化、保護器官功能具有重要作用,因為多數器官損傷均屬於不可逆轉的。延誤治療會顯著降低療效,甚至錯失最佳的治療時機。19,20 因此,及早診斷與介入可延緩疾病進展、最大程度地保護多器官功能及改善長期預後。2,19,20研究顯示,在專科團隊協作下接受適當治療及定期追蹤病情,大部分患者的臨床症狀與生活品質均有顯著改善。1,2,19
此專欄由武田藥品全力支持,資料由受訪者提供。內容僅供學術分享、疾病認知或參考之用,並不構成任何醫療建議或藥物推廣,亦不代表其他醫生或醫護專業人員之意見。有關您的個人健康或治療情況,請向您的醫護人員查詢
參考資料:
1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
2. Ortiz A. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123:416-427.
3. Eng CM, et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184-192.
4. Chien Y-H, et al. Fabry Disease: Incidence of the Common Later-Onset α-Galactosidase A IVS4+919G→A Mutation in Taiwanese Newborns—Superiority of DNA-Based to Enzyme-Based Newborn Screening for Common Mutations. Mol Med. 2012;18:780-784.
5. Leung SPY, et al. The Asian Fabry Cardiomyopathy High-Risk Screening Study 2 (ASIAN-FAME-2): Prevalence of Fabry Disease in Patients with Left Ventricular Hypertrophy. J Clin Med. 2024;13:3896.
6. Ishii S. Alternative Splicing in the α-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am J Hum Genet. 2002;70:994-1002.
7. Suppah M. Fabry’s Disease as a Potential Overlapping Diagnosis in Progressive Hypertrophic Cardiomyopathy. Abstract 14735. Presented at the American Heart Association Scientific Sessions & Resuscitation Science Symposium; Nov 6, 2023.
8. Lin C-J. Insights of Fabry disease: Expert consensus approach for screening, diagnosis, and multidisciplinary management in chronic kidney disease. J Formos Med Assoc. 2025;124:794-799.
9. Savary AL, et al. Enhancing the diagnosis of Fabry disease in cardiology with a targeted information: a before–after control–impact study. Open Heart. 2017;4:e000567.
10. Kurdi H, et al. Finding Fabry: a survey on missed opportunities for detection and diagnosis of Fabry disease in patients with LVH. Br J Cardiol. 2025;32:26-30.
11. Mehta A, et al. Fabry disease: a review of current management strategies. Q J Med. 2010;103:641659.
12. Rekova P, et al. Missed diagnosis of Fabry disease: should we screen patients with multiple sclerosis? Neurol Sci. 2024;45:231-239.
13. Niemann M, et al. Gene Mutations Versus Clinically Relevant Phenotypes: Lyso-Gb3 Defines Fabry Disease. Circ Cardiovasc Genet. 2014;7:8-16.
14. Duro G, et al. Diagnosis of Fabry Disease Using Alpha-Galactosidase A Activity or LysoGb3 in Blood Fails to Identify Up to Two Thirds of Female Patients. Int J Mol Sci. 2024;25:5158.
15. Nowak A, et al. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol Genet Metab. 2017;120:57-61.
16. Yim J, et al. Fabry Cardiomyopathy: Current Practice and Future Directions. 2021;10:1532.
17. Hospital Authority – HA Drug Formulary. Agalsidase alfa. Available at: https://www.ha.org.hk/hadf/tc/Others/SearchResult.html?text=agalsidase%20alfa#gsc.tab=0. Accessed March 2026.
18. Hospital Authority – HA Drug Formulary. Migalastat. Available at: https://www.ha.org.hk/hadf/tc/Others/SearchResult.html?text=migalastat#gsc.tab=0. Accessed March 2026.
19. Lenders M, et al. Progress and Challenges in the Treatment of Fabry Disease. 2025;39:517-535.
20. Linhart A, et al. An Expert Consensus Document on the Management of Cardiovascular Manifestations of Fabry Disease. Eur J Heart Fail. 2020;22:1076-1096.
C-ANPROM/HK/REP/0024 (06/2026)
熱門文章

2025/05/09

2024/04/18

2024/04/15

2024/04/12

2024/04/12