

身體突然出現水腫以為是過敏?原來反覆出現水腫可能是一種罕見疾病——遺傳性血管性水腫(Hereditary Angioedema;HAE)的症狀之一。1 這種疾病的反覆水腫發作及其遺傳特徵,往往對患者與家人的生活造成了很大困擾。1 因此,及早診斷和有效治療十分重要。1 就讓免疫及過敏病專科醫生李曦醫生帶大家認識這個罕見遺傳病及其近年的治療新突破。
遺傳性血管性水腫是什麼?目前香港的發病率如何?每年大約有多少宗新增確診個案?診斷新病例有什麼困難?
遺傳性血管性水腫是一種罕見遺傳病,主要是由於缺乏C1 抑制因子(C1-inhibitor)或其功能異常所致,屬於補體系統相關的酶抑制因子缺陷。1 由於C1 抑制因子不足,體內血管活性物質(如緩激肽)調控失衡,導致血管通透性增加,從而反覆出現皮膚及黏膜的水腫,通常出現於四肢、面部、上呼吸道及腸道。1
由於遺傳性血管性水腫為常染色體顯性遺傳疾病,患者家族成員罹患風險較高,可透過已確診個案進一步進行家族篩查。1,2 香港目前累積確診個案約72宗,估計發病率為每 16 萬人約有1宗病例,與歐美地區相近;3,4 實際確診數字則取決於各地對疾病的認知及診斷能力。4 新病例的診斷仍具挑戰性,主要原因包括疾病罕見性、臨床表現多樣且容易與過敏性水腫混淆,加上醫護人員及公眾對遺傳性血管性水腫的認知不足,導致不少患者經歷多年反覆發作仍未能確診。5-7 近年隨着醫學教育及疾病意識逐步提升,透過臨床警覺性及家族史追查,可縮短診斷時間,改善新病例的識別與管理。3,5,6
由於遺傳性血管性水腫是遺傳性疾病,若患者確診,是否代表其家族成員也有較高的罹患風險?
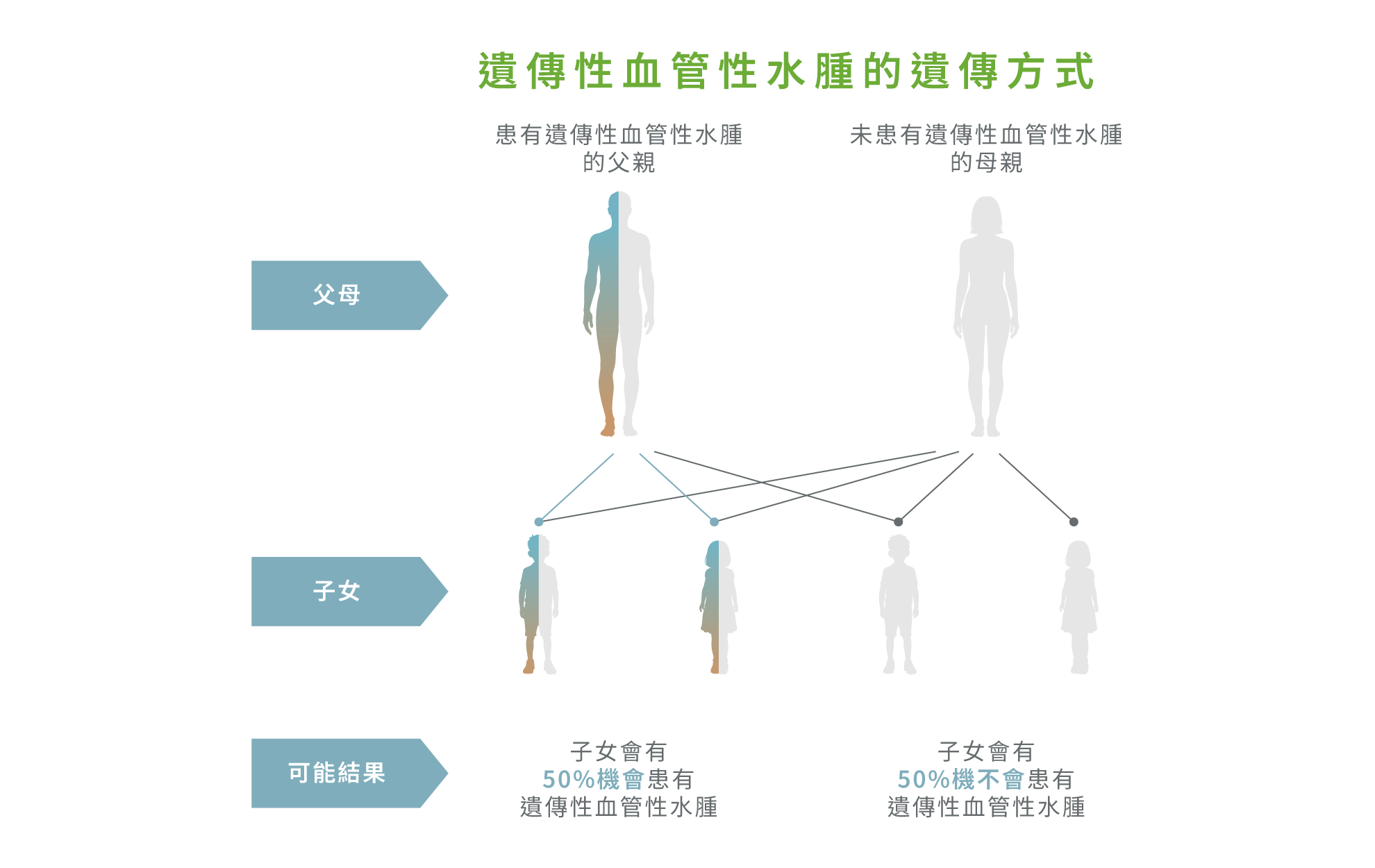
由於遺傳性血管性水腫屬於常染色體顯性遺傳疾病,若患者確診,其一級親屬(如父母、兄弟姊妹及子女)約有 50% 的機會攜帶相關致病基因,罹患風險明顯較一般人高。1,2 臨床觀察顯示,約三分之二以上的患者具有明確的家族病史,且疾病可在同一家族中跨世代出現,並不限於單一分支。1,5,8 部分帶有致病基因的家族成員即使暫時沒有明顯症狀,仍可能在未來出現急性水腫發作。3
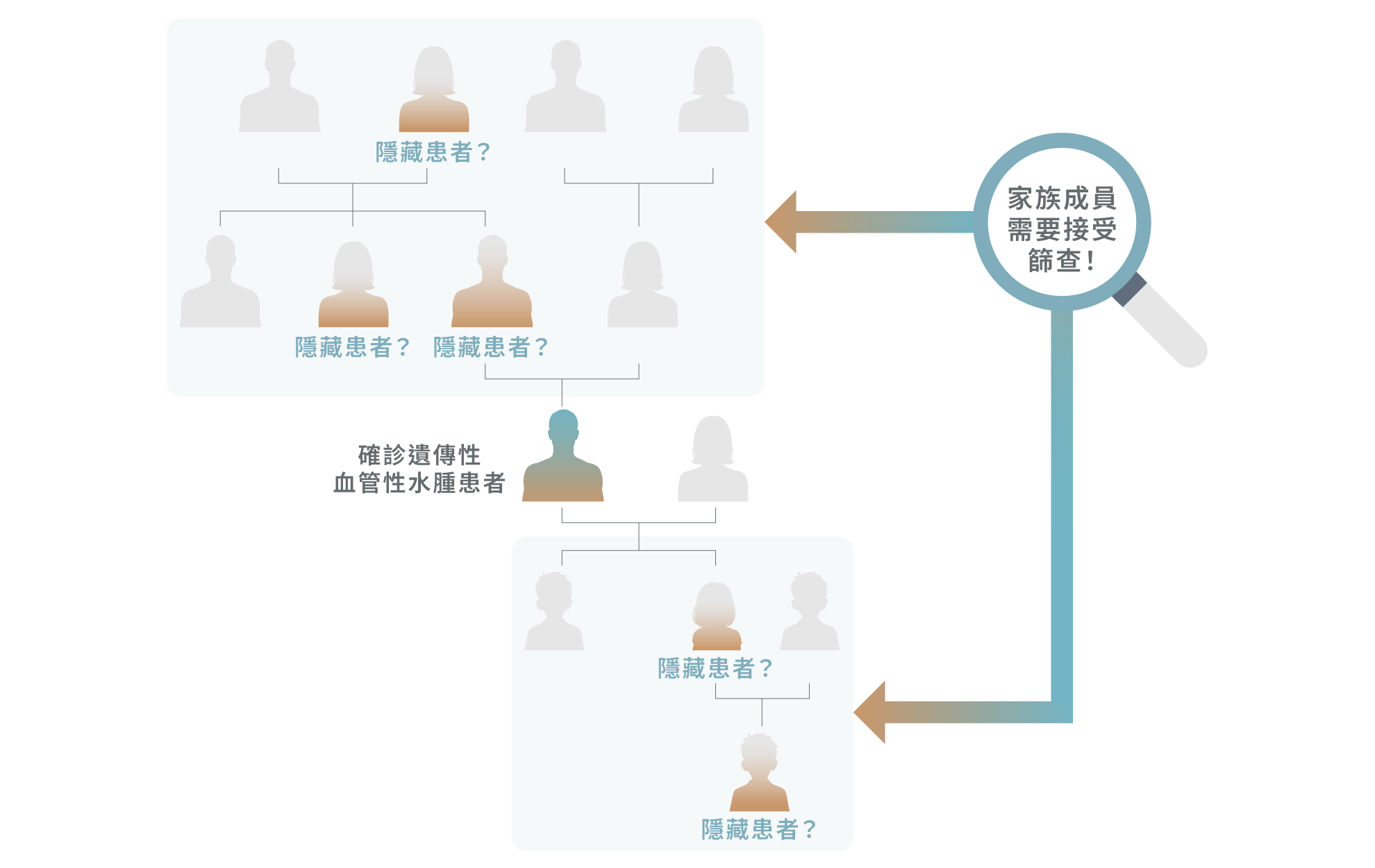
因此,一旦出現確診個案,應進行「家族連鎖篩查」(Cascade family screening),為所有家族成員進行評估與檢測,即使無症狀亦不應忽視。3 及早識別高風險人士,不僅有助預防嚴重急性發作,也對下一代的疾病預防與長遠健康管理具重要意義。3
遺傳性血管性水腫發作時有哪些常見症狀?如何區分這些症狀與一般過敏反應?是否只影響面部或皮膚?
遺傳性血管性水腫常被誤診為一般過敏反應,尤其在疾病初期。不少患者因反覆出現面部或皮膚腫脹而被誤診為過敏性水腫。1,4 然而,與一般過敏反應不同,遺傳性血管性水腫發作時通常沒有伴隨蕁麻疹或明顯痕癢,且對抗組織胺或類固醇治療反應不佳。1,2,5-7 正確認識相關症狀,有助及早分辨遺傳性血管性水腫與過敏反應,減少誤診與延誤治療的風險。2,7
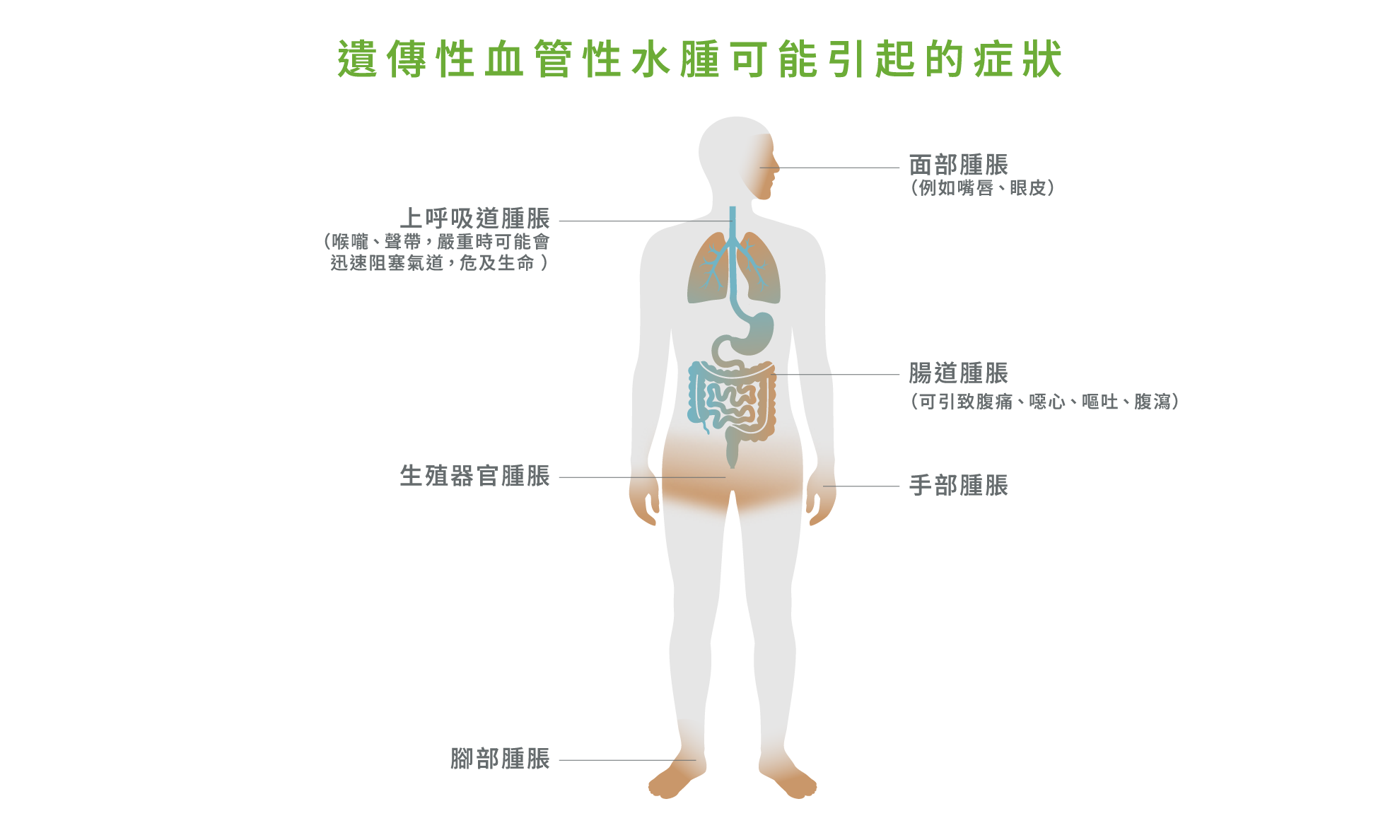
除了面部腫脹,手腳腫脹亦十分常見。有患者曾出現手指突然腫脹至無法配戴戒指,或雙腳腫脹至鞋子不合適。1-3,6,7 部分患者亦可能出現生殖器官腫脹,對日常生活及親密關係造成明顯影響。9 由於疾病可能導致外觀改變,患者亦可能承受社交壓力或被誤解為患上傳染性疾病,影響心理及社交生活。9,10
遺傳性血管性水腫更可能影響腸道,導致劇烈腹痛、嘔吐及腹瀉,臨床表現類似急性腹症,部分患者甚至曾因此接受不必要的手術。1-3,6,7 最危險的情況則為舌頭或上呼吸道腫脹,可能會迅速阻塞氣道,危及生命。1,2,5,7
如何診斷遺傳性血管性水腫?目前香港是否有特定的檢測能夠有效確診此病?
遺傳性血管性水腫的診斷以臨床診斷為關鍵,再結合病史及血液檢測作出綜合評估。1,2,5-7 醫生會留意患者是否反覆出現無明顯痕癢或蕁麻疹的血管性水腫,評估其發作部位、持續時間及是否有家族病史;同時亦需排除其他常見原因,如血管張力素轉換酶抑制劑(ACE inhibitors)所引起的藥物性水腫。1,2,5-7,11

當排除其他常見成因後,如果醫生仍然懷疑是遺傳性血管性水腫,會安排進行C4 水平檢測以作初步篩查。5 若在非發作期發現C4 水平持續偏低,便應進一步進行確診性檢測。1,2,5 現時香港有三種確診性檢測,包括C1抑制蛋白的血濃度測試、C1抑制蛋白功能測試,以及基因檢測。12 當中,C1 抑制因子血濃度及功能測試已可識別大部分個案,而基因檢測則有助於輔助診斷或用於嬰幼兒、產前評估或進行家族篩查。1,2,5 透過結合臨床警覺性與系統化檢測流程,可有效提高遺傳性血管性水腫的確診率,減少誤診及延誤治療的情況。3,5,11
現時香港有哪些治療方案可用於控制遺傳性血管性水腫患者的病情、預防性治療或急性發作管理?目前有哪些藥物是已列入醫管局的藥物名冊供患者使用?

遺傳性血管性水腫的治療目標可分為三個級別:1
- 第一級:迅速控制急性症狀,以降低發作期間的風險
- 第二級:透過疾病控制及預防性治療減少發作頻率與嚴重程度
- 第三級:協助患者回復正常生活節奏,改善整體生活質素
目前香港的治療策略結合了「急性發作管理」及「長期預防性治療」,目標是全面控制症狀,讓患者能回復正常的生活質素。11
在處理急性發作時,醫管局藥物名冊已納入C1抑制因子替代治療,以補充患者體內C1抑制因子。雖然此治療安全性高且適合各年齡層,但因需要經靜脈注射,通常要前往醫療機構進行,對患者的日常生活較為不便。1,6,7,11 另一種已列入名冊的選擇是稱為「急救藥物」的緩激肽B2受體拮抗劑,同樣可用於急性發作,適用於成人及兒童的各類型遺傳性血管性水腫,並可由患者自行進行皮下注射。1,6,7,11 研究及臨床經驗顯示,及早使用緩激肽B2受體拮抗劑有助縮短發作持續時間、減輕症狀嚴重程度,亦能克服即時就醫的障礙,因此現時大部分已確診的遺傳性血管性水腫患者均會隨身攜帶備用。1,6,7
在預防性治療方面,國際指南已將長效預防性治療列為一線選擇之一,其中包括每四星期皮下注射一次的血漿激肽釋放酶抑制劑,亦稱單克隆抗體,能有效降低遺傳性血管性水腫發作的頻率及嚴重程度。1,2,6,7,11 然而,香港相關預防性藥物暫未納入醫管局常規資助藥物名冊,現階段主要透過恩恤用藥(Compassionate Use)或臨床研究途徑提供予合適的患者。11
整體而言,隨着治療選擇日趨多元化,遺傳性血管性水腫的管理已不再局限於應付急性發作,而是朝向長期穩定控制疾病、讓患者重返正常生活及提升生活質素的方向發展。1
雖然遺傳性血管性水腫是一種終身疾病,患者應如何與疾病共存?醫生是否有相關的個案分享或建議,以鼓勵受到此疾病困擾的人士尋求幫助?
雖然遺傳性血管性水腫屬於終身疾病,但隨着醫療科技不斷進步,患者可以透過適切管理與疾病和平共存,並維持正常的生活。2 李醫生強調及早確診、正確認識疾病,以及建立長期治療與應變計劃,對穩定病情及提升生活質素至關重要。1,6
李醫生亦分享了一個個案。曾有一名女性患者因家族中三代均受遺傳性血管性水腫影響,在祖母及母親年代,因缺乏有效治療選擇而長期忍受病情,令她一度對求醫及診斷失去信心,甚至對生育及組織家庭感到絕望。直至近年新型藥物出現,在醫生詳細解釋及支援下,她逐漸接受診斷並開始接受治療,隨後能夠安心規劃家庭,順利經歷兩次懷孕,充分反映遺傳性血管性水腫並非「無藥可醫」,亦不會成為人生規劃的阻礙。李醫生藉此鼓勵患者及其家人不要因過往經驗而放棄求醫,亦毋須過度擔憂,只要配合治療及定期覆診,大部分患者均可有效控制病情,重返日常生活。2
展望未來,醫護界亦期望「預防性治療」能逐步納入資助範圍,讓更多遺傳性血管性水腫患者受惠,進一步改善整體照護與生活質素。11
此專欄由武田藥品全力支持,資料由受訪者提供。內容僅供學術分享、疾病認知或參考之用,並不構成任何醫療建議或藥物推廣,亦不代表其他醫生或醫護專業人員之意見。有關您的個人健康或治療情況,請向您的醫護人員查詢。
參考資料:
1. Maurer M, et al. The international WAO EAACI guideline for the management of hereditary angioedema: The 2021 revision and update. Allergy. 2022;77:1961-1990.
2. Busse PJ, et al. US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9:132-150.
3. Wong JCY, et al. Prospective study on the efficacy and impact of cascade screening and evaluation of hereditary angioedema (CaSE-HAE). J Allergy Clin Immunol Pract. 2022;10:2896-2903.
4. Li PH, et al. Epidemiology, management, and treatment access of hereditary angioedema in the Asia Pacific region: Outcomes from an international survey. J Allergy Clin Immunol Pract. 2023;11:1253-1260.
5. Grumach AS, et al. Hereditary angioedema diagnosis: Reflecting on the past, envisioning the future. World Allergy Organ J. 2025;18:101060.
6. Lochbaum R, Staubach-Renz P, et al. Treatment of hereditary angioedema: Advances and gaps in care. Allergo J Int. 2025.
7. Rosi-Schumacher M, et al. Clinical manifestations of hereditary angioedema and a systematic review of treatment options. Laryngoscope Investig Otolaryngol. 2021;6:394-403.
8. Zuraw BL, et al. The pathophysiology of hereditary angioedema. World Allergy Organ J. 2010;3(9 Suppl):S25-S28.
9. Camyar A, et al. Genital attacks in hereditary angioedema and their effects on sexual life. Medicina. 2024;60:1777.
10. Valentim J, et al. Experiences of stigmatization and its impacts among individuals living with hereditary diseases and family members in Portugal: An exploratory mixed-method study. J Community Genet. 2025;16(6):861-872.
11. Li PH, et al. Hong Kong-Macau severe hives and angioedema referral pathway. Front Allergy. 2023;4:1290021.
12. Li PH, et al. Hereditary angioedema in China: advancing awareness, access, advocacy and alliances from the Greater Bay Area to the global HAE community. Clin Exp Allergy. 2025;55:659-670.
C-ANPROM/HK/FIR/0022 (04/2026)
熱門文章

2025/05/09

2024/04/18

2024/04/15

2024/04/12

2024/04/12